Secondo ricercatori della Rice University, lo sforzo di sviluppare un software che svela la complessità del ripiegamento delle proteine sta ripagando con nuove conoscenze su questo fondamentale processo.
Lo studio pubblicato questa settimana nei Proceedings of the National Academy of Sciences dal chimico Peter Wolynes e dal suo team di ricerca della BioScience Research Collaborative della Rice, dovrebbe essere di particolare interesse per coloro che sondano le radici delle malattie degenerative associate all'aggregazione di fibre amiloidi, come Alzheimer, Parkinson e diabete di tipo 2.
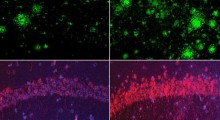
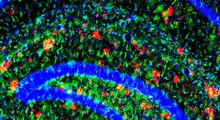
 Un grafico tridimensionale delle energie all'opera nel ripiegamento delle proteine in multidominio, prodotto alla Rice University con il programma AWSEM-MD, mostra le regioni stabili in blu nella parte inferiore ("N") della proteina nel suo stato nativo corretto, e nella parte superiore ( "I") nel suo stato mal-ripiegato errato. Entrambi gli estremi sono ugualmente stabili, anche se le proteine "N" hanno meno energia. Rosso e verde rappresentano i domini proteici separati; i colori sono mescolati nella struttura con dominio scambiato (in alto) e nella struttura di "auto-riconoscimento" (a destra), che si sono mal ripiegate a causa di forti interazioni di auto-riconoscimento tra le sequenze di residui brevi mostrate nelle strisce colorate. I ricercatori sospettano che le proteine mal ripiegate abbiano un ruolo importante nella aggregazione delle fibre amiloidi implicate nelle malattie degenerative. (Credit: Image by Nicholas Schafer and Weihua Zheng) |
Il software di dinamica molecolare, idoneo a prevedere come si curvano e si avvolgono nelle varie forme funzionali i filamenti dei residui, è stato progettato per seguire l'innovativo "principio di minima frustrazione" di Wolynes e colleghi. Questi residui, i granelli molecolari che costituiscono le proteine, mentre si piegano nel loro stato di origine, seguono il percorso di minor resistenza. Il principio descrive il modo in cui l'evoluzione ha plasmato il percorso che segue una proteina verso la stabilità.
Il software, chiamato AWSEM-MD (Associative memory, Water-mediated Structure and Energy Model) simula i possibili modi in cui si dovrebbero piegare i granelli di una striscia, sulla base delle energie in gioco, fino al livello submolecolare, e predice accuratamente la struttura finale. Due sviluppatori della versione corrente, Weihua Zheng, ricercatore post-dottorato alla Rice, e Nicholas Schafer, studente laureato, sono co-autori del nuovo studio, l'ultimo di una serie sulle dinamiche di piegatura definite da software.
I ricercatori si proponevano di confermare che un processo, visto da sperimentatori e chiamato scambio di dominio, è una delle cause del misfolding [=errata piegatura delle proteine]. I domini sono parti di catene proteiche conservate. A volte un dominio di una catena può incontrare il suo sosia di una catena vicina e impigliarsi con esso tramite interazioni simili a quelle dello stato di piegatura corretta. Il risultato è spesso un dimero - proteine come gemelli siamesi - che non sarà probabilmente in grado di svolgere il compito biologico previsto e può diventare parte di una fibrilla amiloide dannosa. "Gli sperimentatori hanno avuto qualche evidenza forte in laboratorio che la dimerizzazione è una conseguenza della minima frustrazione, un'idea proposta in precedenza dal nostro gruppo su un piano più generale", ha detto Wolynes. "Quindi abbiamo pensato che sarebbe stato bene fare una simulazione per controllarlo".
Il team ha visto effettivamente lo scambio di dominio nei loro modelli di titina cardiaca umana, una proteina muscolare. Ma sono stati sorpresi di vedere qualcosa di inaspettato: la prova che sequenze identiche di catene vicine, composte di 5-7 residui solamente, hanno la spiacevole tendenza di incontrarsi e stare insieme. Hanno trovato istanze che tale "auto-riconoscimento" fa pendere la bilancia delle energie che decide se una proteina si può piegare correttamente. Sostituendo anche solo alcuni residui di un frammento, si elimina l'auto-riconoscimento e si abbassa l'incidenza degli scambi di dominio, dice Wolynes.
|
|
"Non eravamo persone che pensavano a questa possibilità", ha detto. "Era stata suggerita da altri, anche se non ci avevo mai creduto, perché non c'è una connessione evidente con il principio della frustrazione minima". Ma le simulazioni mostrano casi dove l'auto-riconoscimento appiccicoso di un segmento in una catena potrebbe influenzare l'energia dei residui della serie e instaurare efficacemente una "frustrazione" che impedisce al resto della proteina di piegarsi del tutto, con il risultato di grande disordine, o entropia.
Anche se i modelli non si collegano direttamente alla formazione di fibrille amiloidi, dice Wolynes, l'aneddotica indica che le malattie da ripiegamento delle proteine hanno una certa correlazione con la febbre che permette all'entropia aggiuntiva di stabilizzare le forme mal ripegate. "I risultati forniscono una nuova spiegazione", dice, "di come una parte disordinata della catena può contribuire alla stabilità di questi stati mal ripiegati ad alta temperatura". "Quando ti dice 'prendi due aspirine e chiamami domattina', il medico ti sta facendo un favore più grande di quello che credi", dice Wolynes.
La scoperta potrebbe aprire la strada alla progettazione di farmaci che inibiscono specifiche interazioni. "Nella simulazione al computer, questo auto-riconoscimento sembra essere impedito da modifiche minime, e questo è quello che vogliamo che facciano gli sperimentatori: modifiche per vedere se diminuisce l'effetto auto-riconoscimento", ha detto. "Le nostre simulazioni forniscono dettagli strutturali di proteine mal ripiegate a livello molecolare, che sono difficili da indagare negli esperimenti", ha detto Zheng. "Queste possono generare ipotesi specifiche da testare".
I ricercatori sperano che il loro lavoro sarà utile sia agli sperimentatori che ad altri ricercatori computazionali sulla piegatura delle proteine. "L'AWSEM è ospitato su Google Code, cosa che richiede tutto codice open-source", dice Schafer. "Quindi è a disposizione di chiunque voglia usarlo. Quello che stiamo vedendo con questi studi è che i valori che otteniamo applicando il principio di frustrazione minima sono appropriati a livello mondiale, non solo per predire le strutture native delle proteine. Può prevedere pure le strutture associate (come i dimeri) e le strutture mal ripiegate. Bisogna sempre stare attenti a utilizzare modelli che 'maneggiano il mazzo' a favore di un determinato risultato atteso", dice. "Ma ciò che è interessante è che il nostro modello non dispone di informazioni a priori su questi specifici tipi di strutture mal ripiegate. Il nostro modello è parametrizzato usando come input solo i dati sperimentali delle strutture piegate correttamente e quindi applicando il principio di frustrazione minima. L'ampia gamma di successi che abbiamo avuto quest'anno ci dice che abbiamo un metodo decente per ricavare i punti di forza delle interazioni".
"Non abbiamo mai veramente pensato a specifici tipi di misfolding o al processo di aggregazione quando abbiamo costruito il nostro modello intorno al principio di frustrazione minima", dice Zheng. "Ma sono tutti a posto".
La versione corrente di AWSEM è stata progettata e programmata per la maggior parte da Garegin Papoian, Professore Associato "Monroe Martin" dell'Università del Maryland, e da Aram Davtyan, studente laureato del suo laboratorio; questa versione si basa sullo sviluppo precedente di Cecilia Clementi, docente "Wiess Career Development" e professore di chimica e di ingegneria chimica e biomolecolare alla Rice, e altri del laboratorio di Wolynes all'Università della California di San Diego e alla University of Illinois di Urbana-Champaign.
***********************
Cosa pensi di questo articolo? Ti è stato utile? Hai rilievi, riserve, integrazioni? Conosci casi o ti è successo qualcosa che lo conferma? o lo smentisce? Puoi usare il modulo dei commenti qui sotto per dire la tua opinione. Che è importante e unica.
***********************
Fonte: Materiale della Rice University. Articolo originale scritto da Mike Williams.
Riferimento: Weihua Zheng, Nicholas P. Schafer, and Peter G. Wolynes. Frustration in the energy landscapes of multidomain protein misfolding. PNAS, January 14, 2013 DOI: 10.1073/pnas.1222130110.
Pubblicato in ScienceDaily il 14 Gennaio 2013 - Traduzione di Franco Pellizzari.
Copyright: Tutti i diritti di eventuali testi o marchi citati nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non dipende da, nè impegna l'Associazione Alzheimer onlus di Riese Pio X. I siti terzi raggiungibili da eventuali links contenuti nell'articolo e/o dagli annunci pubblicitari proposti da Google sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.
| Sostieni l'Associazione; una donazione, anche minima, ci aiuterà ad assistere malati e famiglie e continuare ad informarti. Clicca qui a destra: |