Puntare il percorso di segnalazione appena identificato è promettente per trattare i disturbi neurodegenerativi.
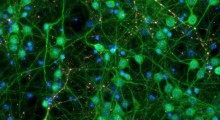
.") A. Localizzazione dei componenti di segnalazione del percorso WNT/PCP nelle sinapsi glutammatergiche dell'ippocampo adulto, rilevati dalla microscopia a super risoluzione.
A. Localizzazione dei componenti di segnalazione del percorso WNT/PCP nelle sinapsi glutammatergiche dell'ippocampo adulto, rilevati dalla microscopia a super risoluzione.
B. Diagramma schematico che mostra il bilanciamento della segnalazione WNT/PCP nella manutenzione delle sinapsi e il sito di legame dell'amiloide-beta oligomerica. (Fonte: Feng et al.).
Il cervello degli adulti sani è dotato di un vasto numero di sinapsi, strutture che permettono il passaggio dei segnali tra le cellule nervose per abilitare la comunicazione, l'elaborazione e la conservazione delle informazioni in tutto il sistema nervoso.
A parte i periodi dinamici, quando il cervello sta apprendendo nuove informazioni o abilità, il numero delle sinapsi 'glutammatergiche', il principale tipo di sinapsi che usano i neuroni per attivarsi l'uno con l'altro, rimane in gran parte costante negli adulti.
Nei disordini del cervello come il morbo di Alzheimer (MA), queste connessioni sinaptiche, che contengono i nostri preziosi ricordi, sono note per rompersi troppo presto e scomparire. Si ritiene che questa degenerazione delle sinapsi inizi molto prima della perdita della memoria e che acceleri con la progressione della malattia.
Le cause della degenerazione delle sinapsi nei disturbi neurodegenerativi non sono ancora ben comprese, principalmente perché gli scienziati non hanno ancora svelato i meccanismi chiave che di norma tengono insieme queste strutture minuscole (mediamente un micrometro di diametro) per tutta la vita.
Dei neurobiologi dell'Università della California di San Diego hanno ora scoperto i meccanismi a lungo ricercati dietro il mantenimento delle sinapsi glutammatergiche. In base a questa scoperta fondamentale, il ricercatore post-dottorato della divisione di scienze biologiche Bo Feng, il prof. Yimin Zou, e i loro colleghi, hanno identificato i componenti principali che guidano la degenerazione della sinapsi associate all'amiloide-beta (Aβ). L'Aβ è formata da peptidi di 36-43 amminoacidi derivati dalla proteina precursore dell'amiloide (APP) ed è il componente principale delle placche amiloidi presenti nel cervello di persone con MA.
Nonostante sforzi tremendi, non è stato ancora possibile scoprire un farmaco per il MA. Finora, gli approcci principali sono stati ridurre la produzione di Aβ o eliminare le placche che essa forma. La nuova scoperta dei ricercatori dell'UC San Diego, pubblicata su Science Advances il 18 agosto 2021, suggerisce un approccio alternativo più a valle: proteggere le sinapsi bloccando direttamente le azioni tossiche dell'Aβ.
Le sinapsi glutammatergiche sono strutture altamente polarizzate con una parte pre-sinaptica da una cellula nervosa e una parte post-sinaptica da un'altra. Questo tipo di polarità garantisce la corretta direzione del flusso di informazioni. Il laboratorio di Zou aveva in precedenza scoperto che durante lo sviluppo del cervello le strutture sinaptiche altamente polarizzate sono assemblate da componenti del percorso 'polarità cellulare planare' (PCP, planar cell polarity): un percorso di segnalazione potente che polarizza le giunzioni tra una cellula e l'altra lungo il piano del tessuto.
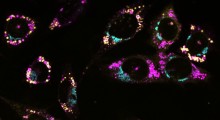
Usando la microscopia a super risoluzione, i ricercatori hanno rilevato la posizione precisa di questi componenti di segnalazione PCP, chiamati Celsr3, Frizzled3 e Vangl2, nelle sinapsi glutammatergiche nel cervello adulto. Hanno quindi scoperto che la rimozione di questi componenti, essenziali per l'assemblaggio iniziale delle sinapsi da parte dei neuroni adulti, può alterare drasticamente il numero di sinapsi.
Queste scoperte sorprendenti suggeriscono che il numero di sinapsi complessivo in un cervello normale viene mantenuto da un buon equilibrio tra Celsr3 (che stabilizza la sinapsi) e Vangl2 (che scompone le sinapsi). Desiderosi di capire se questi componenti sono coinvolti nella degenerazione delle sinapsi, i ricercatori hanno testato se l'Aβ, una guida cruciale della perdita di sinapsi nel MA, influisce sulla funzione o sull'interazione di queste proteine.
In una serie di esperimenti, hanno dimostrato che gli oligomeri Aβ si legano a Celsr3 e consentono a Vangl2 di smontare più efficacemente le sinapsi, probabilmente indebolendo le interazioni tra Celsr3 e Frizzled3. "È come se l'Aβ avesse scoperto da tempo il tallone d'Achille delle nostre sinapsi", ha detto Zou, professore nella Sezione di Neurobiologia della Divisione di Scienze Biologiche.
Quando i ricercatori hanno rimosso i Vangl2 dai neuroni, hanno scoperto che l'Aβ non riusciva più a causare la degenerazione delle sinapsi, sia nelle colture neuronali che negli animali esposti agli oligomeri Aβ.
Anche il Ryk, un regolatore del percorso PCP, che interagisce con Frizzled3 e Vangl2, si trova nelle sinapsi adulte e funziona allo stesso modo dei Vangl2 per mediare lo smontaggio della sinapsi. I ricercatori hanno scoperto che bloccare il Ryk con anticorpi di blocco delle funzioni può proteggere le sinapsi dalla degenerazione indotta da Aβ.
Per verificare ulteriormente l'ipotesi se questo percorso di segnalazione fondamentale è un obiettivo primario della degenerazione delle sinapsi nel MA, il laboratorio di Zou ha usato topi 5xfad, un modello della patologia Aβ. Questo topo transgenico porta cinque mutazioni umane che causano il MA e quindi mostra i sintomi gravi della degenerazione delle sinapsi e la perdita di funzione cognitiva.
Hanno scoperto che la rimozione del Ryk con la soppressione genetica dai neuroni adulti proteggeva le sinapsi e conservava la funzione cognitiva dei topi 5xfad. L'infusione dell'anticorpo che blocca la funzione del Ryk ha anche protetto le sinapsi e ha conservato la funzione cognitiva nei topi 5xFad, suggerendo che l'anticorpo Ryk è un potenziale agente terapeutico.
Questi risultati entusiasmanti suggeriscono che il percorso PCP è un obiettivo diretto della perdita di sinapsi indotta da Aβ nel MA.
"Poiché la patologia Aβ e la perdita di sinapsi di solito si verifica nelle prime fasi del MA, anche prima che sia evidente il declino cognitivo, l'intervento precoce, come il ripristino del riequilibrio del percorso PCP, darà probabilmente dei vantaggi ai pazienti di MA", ha detto Zou.
Anche la neuroinfiammazione, riflessa dall'attivazione degli astrociti e delle microglia, è un segno distintivo della patologia di MA, che può essere indotta dall'accumulo di Aβ ed è nota per accelerare la perdita di sinapsi. È interessante che il laboratorio di Zou abbia scoperto che l'anticorpo Ryk può anche bloccare l'attivazione di astrociti e microglia nei topi 5xfad.
Sebbene non possano distinguere se ciò sia dovuto all'effetto indiretto della protezione delle sinapsi o al blocco delle funzioni Ryk nell'infiammazione, o per entrambi, Zou ritiene che i risultati siano coerenti con il miglioramento del comportamento cognitivo e supporta ulteriormente il Ryk come potenziale bersaglio terapeutico sia per proteggere le sinapsi che per ridurre l'infiammazione nel MA.
"Questa scoperta può essere applicabile alla degenerazione delle sinapsi in generale, poiché i componenti PCP possono essere gli obiettivi sinaptici diretti che mediano la perdita di sinapsi in altri disturbi neurodegenerativi, come il Parkinson e la sclerosi laterale amiotrofica", conclude Zou.
Fonte: Mario Aguilera in University of California - San Diego (> English) - Traduzione di Franco Pellizzari.
Riferimenti: Bo Feng, Andiara Freitas, Lilach Gorodetski, Jingyi Wang, Runyi Tian, Yeo Rang Lee, Akumbir Grewal, Yimin Zou. Planar cell polarity signaling components are a direct target of β-amyloid–associated degeneration of glutamatergic synapses. Science Advances, 2021, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.