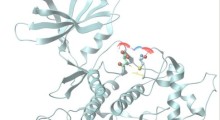
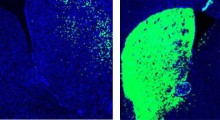
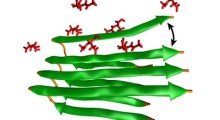
Schema di propagazione dei prioni
Schema di propagazione dei prioni
Ricercatori della Case Western Reserve University che studiano i prioni (proteine mal ripiegate che causano malattie letali incurabili) hanno identificato per la prima volta le caratteristiche della superficie dei prioni umani responsabili della loro replica nel cervello. L'obiettivo finale della ricerca è contribuire a progettare una strategia per fermare le malattie dei prioni negli umani, e, in definitiva, per tradurre nuovi approcci che funzionano sul morbo di Alzheimer (MA) e su altre malattie neurodegenerative.
Gli scienziati devono ancora scoprire la causa esatta del MA, ma in gran parte concordano che i problemi proteici hanno un ruolo nella sua comparsa e progressione. Il MA affligge più di 6 milioni di persone negli Stati Uniti, e l'Alzheimer's Association stima che la loro cura costerà circa $ 355 miliardi quest'anno.
La ricerca è stata fatta al Laboratorio Safar nel Dipartimento di Patologia e nel Centro Proteomica e Bioinformatica alla Case Reserve Western Reserve University, e nel Center for Synchrotron Bioscience dei Brookhaven Laboratories di New York, della stessa università. Safar Jiri, professore di patologia, neurologia e neuroscienze nella Facoltà di Medicina della Case Western Reserve, ha guidato la ricerca, pubblicata il 17 giugno su PLOS Pathogens.
I prioni sono stati scoperti per la prima volta alla fine degli anni '80 come agente biologico contenente proteine che poteva auto replicarsi nelle cellule viventi senza acido nucleico. L'impatto sulla salute pubblica delle malattie del prioni umani trasmesse medicalmente - e anche le trasmissioni animali dell'encefalopatia spongiforme bovina ('malattia della mucca pazza') - ha drasticamente accelerato lo sviluppo di un nuovo concetto scientifico di proteine auto-replicanti.
I prioni umani possono legarsi alla proteine normali vicine nel cervello e causare fori microscopici. In sostanza, trasformano il cervello in strutture spugnose e portano alla demenza e alla morte. Queste scoperte hanno portato al dibattito scientifico in corso sul fatto che i meccanismi di tipo prionico possano essere coinvolti nell'origine e nella diffusione di altri disturbi neurodegenerativi negli esseri umani.
"Le malattie umane da prioni sono plausibilmente i disturbi neurodegenerativi più eterogenei, e un crescente corpo di ricerca indica che sono causati da ceppi distinti di prioni umani", ha detto Safar. "Tuttavia, gli studi strutturali dei prioni umani sono rimasti indietro rispetto ai recenti progressi nei prioni dei topi in laboratorio, in parte a causa delle loro complesse caratteristiche molecolari e dei proibitivi requisiti di biosicurezza richiesti per indagare la malattia, che è invariabilmente fatale e non ha alcun trattamento".
I ricercatori hanno sviluppato un nuovo processo in tre fasi per studiare i prioni umani:
- I prioni derivati dal cervello umano sono stati prima esposti a raggi-X di sincrotrone ad alta intensità. Quel raggio ha creato specie di radicali idrossili che, con brevi lampi di luce, hanno modificato selettivamente e progressivamente la composizione chimica della superficie del prioni. Le proprietà uniche di questo tipo di fonte di luce includono la sua enorme intensità; può essere milioni di volte più luminosa della luce del sole sulla terra.
- Le rapide modifiche chimiche dei prioni causate dai brevi lampi di luce sono state monitorate con anticorpi anti-prionici. Gli anticorpi riconoscono le caratteristiche della superficie del prione e la spettrometria di massa identifica i siti esatti delle differenze specifiche per prione, fornendo una descrizione ancora più precisa dei difetti del prioni.
- Ai prioni illuminati è stato permesso quindi di replicarsi in provetta. La perdita progressiva della loro attività di replicazione mentre il sincrotrone li modificava, ha aiutato a identificare gli elementi strutturali chiave responsabili della replica e della propagazione dei prioni nel cervello.
"Il lavoro è un primo passo fondamentale per identificare i siti di importanza strutturale che riflettono le differenze tra i prioni di diverse diagnosi e aggressività", ha dichiarato Mark Chance, vicepreside per la ricerca e coautore senior del lavoro. "Pertanto, ora possiamo immaginare di progettare piccole molecole che si legano a questi siti di nucleazione e replicazione, e bloccare la progressione delle malattie umane da prioni nei pazienti".
Questo approccio strutturale, ha detto Chance, fornisce anche un modello per identificare i siti strutturalmente importanti su proteine mal ripiegate in altre malattie, come l'Alzheimer, che vedono la propagazione delle proteine da cellula a cellula come fanno i prioni.
Fonte: Case Western Reserve University (> English) - Traduzione di Franco Pellizzari.
Riferimenti: Mohammad Khursheed Siddiqi, Chae Kim, Tracy Haldiman, Miroslava Kacirova, Benlian Wang, Jen Bohon, Mark Chance, Janna Kiselar, Jiri Safar. Structurally distinct external solvent-exposed domains drive replication of major human prions. PLOS Pathogens, 2021, DOI
Copyright: Tutti i diritti di testi o marchi inclusi nell'articolo sono riservati ai rispettivi proprietari.
Liberatoria: Questo articolo non propone terapie o diete; per qualsiasi modifica della propria cura o regime alimentare si consiglia di rivolgersi a un medico o dietologo. Il contenuto non rappresenta necessariamente l'opinione dell'Associazione Alzheimer OdV di Riese Pio X ma solo quella dell'autore citato come "Fonte". I siti terzi raggiungibili da eventuali collegamenti contenuti nell'articolo e/o dagli annunci pubblicitari sono completamente estranei all'Associazione, il loro accesso e uso è a discrezione dell'utente. Liberatoria completa qui.
Nota: L'articolo potrebbe riferire risultati di ricerche mediche, psicologiche, scientifiche o sportive che riflettono lo stato delle conoscenze raggiunte fino alla data della loro pubblicazione.